Clara Parabricks
GCATemplates available: no
Clara Parabricks homepage
module spider parabricks
Clara Parabricks is a software suite that allows users to perform secondary analysis of next generation sequencing DNA and RNA data. The software incorporated into Clara Parabricks has been optimized to run on GPUs, drastically increasing the speed at which common NGS analyses and tasks can be completed.
Examples
Example 1: Complete germline analysis
#!/bin/bash
#SBATCH --job-name=parabricks-germline # set the job name to "parabricks-germline"
#SBATCH --time=02:00:00 # set the wall clock limit to 2 hours
#SBATCH --ntasks-per-node=1 # request one task per node
#SBATCH --cpus-per-task=48 # request 48 cpus per task
#SBATCH --mem=360G # request 360G of memory
#SBATCH --output=%x.%j.stdout # set standard output to write to <jobname>.<jobID>.stdout
#SBATCH --error=%x.%j.stderr # set standard error to write to <jobname>.<jobID>.stderr
#SBATCH --partition=gpu # request the gpu partition
#SBATCH --gres=gpu:h100:2 # request to h100 GPUs
# environment setup
module purge # ensure the working environment is clean
module load parabricks/4.5.0 # load the parabricks module
# run the parabricks germline command
pbrun germline --ref /path/to/indexed/genome.fasta \
--in-fq /path/to/forward_reads.fq /path/to/reverse_reads.fq \
--knownSites /path/to/known/variants.vcf.gz \
--out-bam output.bam \
--out-variants output.vcf \
--out-recal-file output.txt \
--num-gpus 2
#!/bin/bash
#SBATCH --job-name=parabricks-germline # set the job name to "parabricks-germline"
#SBATCH --time=02:00:00 # set the wall clock limit to 2 hours
#SBATCH --ntasks-per-node=1 # request one task per node
#SBATCH --cpus-per-task=64 # request 64 cpus per task
#SBATCH --mem=250G # request 250G of memory
#SBATCH --output=%x.%j.stdout # set standard output to write to <jobname>.<jobID>.stdout
#SBATCH --error=%x.%j.stderr # set standard error to write to <jobname>.<jobID>.stderr
#SBATCH --partition=gpu # request the gpu partition
#SBATCH --gres=gpu:a100:2 # request two A100 GPUs
# environment setup
module purge # ensure the working environment is clean
module load parabricks/4.0.1 # load the parabricks module
# run the parabricks germline command
pbrun germline --ref /path/to/indexed/genome.fasta \
--in-fq /path/to/forward_reads.fq /path/to/reverse_reads.fq \
--knownSites /path/to/known/variants.vcf.gz \
--out-bam output.bam \
--out-variants output.vcf \
--out-recal-file output.txt \
--num-gpus 2
#!/bin/bash
#SBATCH --job-name=parabricks-germline # set the job name to "parabricks-germline"
#SBATCH --time=02:00:00 # set the wall clock limit to 2 hours
#SBATCH --ntasks-per-node=1 # request one task per node
#SBATCH --cpus-per-task=96 # request 94 cpus per task
#SBATCH --mem=488G # request 488G of memory
#SBATCH --output=%x.%j.stdout # set standard output to write to <jobname>.<jobID>.stdout
#SBATCH --error=%x.%j.stderr # set standard error to write to <jobname>.<jobID>.stderr
#SBATCH --partition=gpu # request the gpu partition
#SBATCH --gres=gpu:a100:2 # request two A100 GPUs
# environment setup
module purge # ensure the working environment is clean
module load parabricks/4.1.1 # load the parabricks module
# run the parabricks germline command
pbrun germline --ref /path/to/indexed/genome.fasta \
--in-fq /path/to/forward_reads.fq /path/to/reverse_reads.fq \
--knownSites /path/to/known/variants.vcf.gz \
--out-bam output.bam \
--out-variants output.vcf \
--out-recal-file output.txt \
--num-gpus 2
Example 2: Aligning paired-end RNA-seq data to a genome
Index genomes before running the script with STAR 2.7.2b:
module spider STAR/2.7.2b
Then run the job script:
#!/bin/bash
#SBATCH --job-name=parabricks-rna # set the job name to "parabricks-rna"
#SBATCH --time=01:00:00 # set the wall clock limit to 1 hour
#SBATCH --ntasks-per-node=1 # request one task per node
#SBATCH --cpus-per-task=48 # request 48 cpus per task
#SBATCH --mem=360G # request 360G of memory
#SBATCH --output=%x.%j.stdout # set standard output to write to `<jobname>`.`<jobID>`.stdout
#SBATCH --error=%x.%j.stderr # set standard error to write to `<jobname>`.`<jobID>`.stderr
#SBATCH --partition=gpu # request the gpu partition
#SBATCH --gres=gpu:h100:2 # request two h100 GPUs
# environment setup
module purge # ensure the working environment is clean
module load parabricks/4.5.0 # load the parabricks module
pbrun rna_fq2bam \
--ref /path/to/indexed/genome.fasta \
--genome-lib-dir /path/to/indexed/genome/directory \
--output-dir ./
--in-fq /path/to/forward_reads.fq /path/to/reverse_reads.fq \
--out-bam output.bam \
--num-gpus 2 \
--out-prefix
#!/bin/bash
#SBATCH --job-name=parabricks-rna # set the job name to "parabricks-rna"
#SBATCH --time=01:00:00 # set the wall clock limit to 1 hour
#SBATCH --ntasks-per-node=1 # request one task per node
#SBATCH --cpus-per-task=64 # request 64 cpus per task
#SBATCH --mem=250G # request 250G of memory
#SBATCH --output=%x.%j.stdout # set standard output to write to `<jobname>`.`<jobID>`.stdout
#SBATCH --error=%x.%j.stderr # set standard error to write to `<jobname>`.`<jobID>`.stderr
#SBATCH --partition=gpu # request the gpu partition
#SBATCH --gres=gpu:a100:2 # request two T4 GPUs
# environment setup
module purge # ensure the working environment is clean
module load parabricks/4.0.0 # load the parabricks module
pbrun rna_fq2bam \
--ref /path/to/indexed/genome.fasta \
--genome-lib-dir /path/to/indexed/genome/directory \
--output-dir ./
--in-fq /path/to/forward_reads.fq /path/to/reverse_reads.fq \
--out-bam output.bam \
--num-gpus 2 \
--out-prefix
#!/bin/bash
#SBATCH --job-name=parabricks-rna # set the job name to "parabricks-rna"
#SBATCH --time=01:00:00 # set the wall clock limit to 1 hour
#SBATCH --ntasks-per-node=1 # request one task per node
#SBATCH --cpus-per-task=48 # request 96 cpus per task
#SBATCH --mem=488G # request 488G of memory
#SBATCH --output=%x.%j.stdout # set standard output to write to `<jobname>`.`<jobID>`.stdout
#SBATCH --error=%x.%j.stderr # set standard error to write to `<jobname>`.`<jobID>`.stderr
#SBATCH --partition=gpu # request the gpu partition
#SBATCH --gres=gpu:a100:2 # request two A100 GPUs
# environment setup
module purge # ensure the working environment is clean
module load parabricks/4.0.1 # load the parabricks module
pbrun rna_fq2bam \
--ref /path/to/indexed/genome.fasta \
--genome-lib-dir /path/to/indexed/genome/directory \
--output-dir ./
--in-fq /path/to/forward_reads.fq /path/to/reverse_reads.fq \
--out-bam output.bam \
--num-gpus 2 \
--out-prefix
Using Drona Workflow Engine to run Parabricks Jobs
Drona Workflow Engine, developed by HPRC, provides a 100% graphical interface to generate and submit Parabricks jobs without the need to write a Slurm script yourself or even be aware of Slurm syntax and Parabricks internals. The Drona app is available on all HPRC Portals under the Jobs tab (Screenshot) .
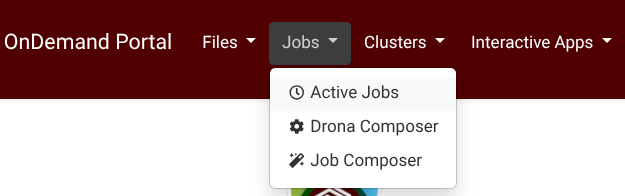{kind=link}
you will find Parabricks in the Environments Dropdown (Screenshot). NOTE: If you don't see Parabricks in the Environments Dropdown, you need to import it first. Click on the + sign next to the environments dropdown and select the Parabricks environment in the pop-up window. You only need to do this once. See the import section for more information about environments.
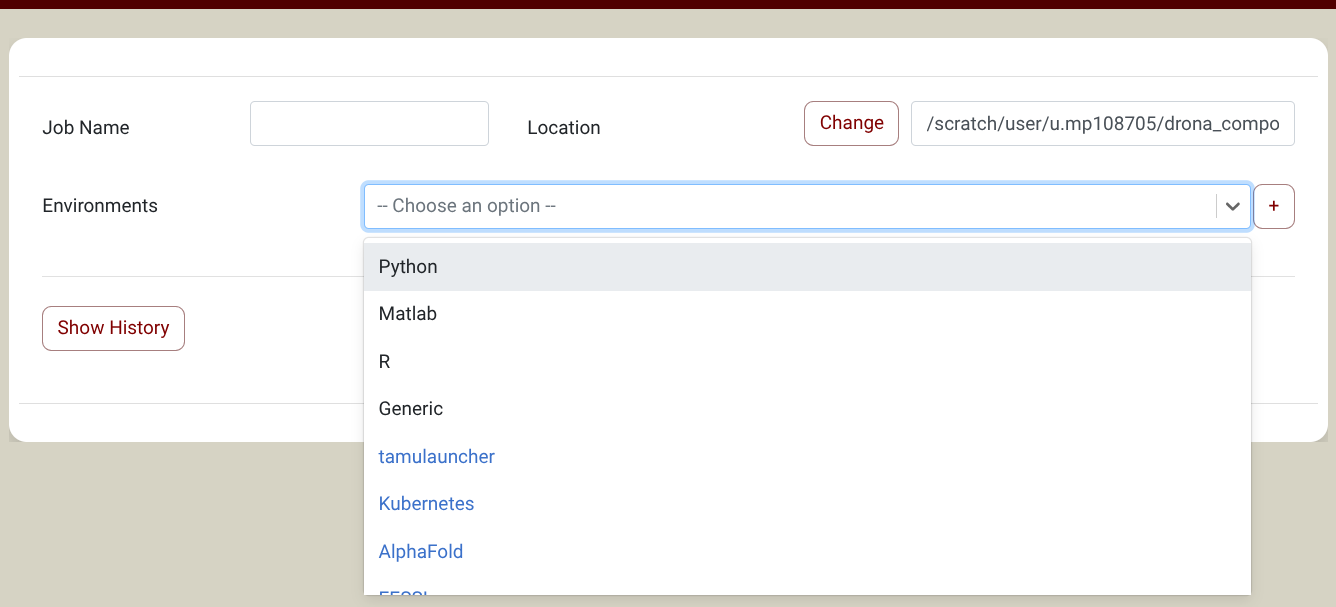{kind=link}
Once you select the Parabricks environment, the form will expand with Parabricks specific fields (Screenshot) to guide you in providing all the needed information. To generate the Parabricks job files, click the Generate or Preview button. This will first show a fully editable preview screen with the generated job scripts. You are welcome to inspect the generated files and make edits if needed. To submit the job, click on the submit button, and Drona will submit the generated job on your behalf. For detailed information about Drona Workflow Engine, checkout the Drona Workflow Engine Guide
{kind=link}
If you experience any issues or have any suggestions, please get in touch with us at [email protected]