LAMMPS
Description
LAMMPS is a classical molecular dynamics code, and an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator from Sandia National Laboratories. - Homepage: http://lammps.sandia.gov/
Documentation: http://lammps.sandia.gov/doc/Manual.html
LAMMPS Acceleration
LAMMPS is commonly built to be accelerated, e.g. distribute computational work in parallel on HPC hardware. There are two main Acceleration Frameworks:
- Kokkos Package provides acceleration for
- NVIDIA GPUs (but not CPUs)
- CPUs
- GPU Package provides acceleration for
- NVIDIA GPUs and CPUs together
- Intel GPUs and CPUs together
NVIDIA GPUs on HPRC clusters:
- T4 on Grace and FASTER
- A100 on Grace and FASTER
- A[10-40] on FASTER, Launch, and ACES
- H100 on ACES
- GH200 on ACES
Intel GPUs on HPRC clusters:
Read more about LAMMPS performance in our selected publications.
LAMMPS Software
LAMMPS is open to all HPRC users.
LAMMPS Modules
NOTE: HPRC typically only installs the stable releases of LAMMPS on the clusters.
To find what LAMMPS versions are available, use module spider:
module spider LAMMPS
To learn how to load a specific module version, use module spider:
module spider LAMMPS/29Sep2021-kokkos
You will need to load all module(s) on any one of the lines below before the "LAMMPS/29Sep2021-kokkos" module is available to load.
GCC/10.2.0 OpenMPI/4.0.5
Read more about toolchains.
Finally, to load LAMMPS:
module load GCC/10.2.0 OpenMPI/4.0.5 LAMMPS/29Sep2021-kokkos
To find what LAMMPS versions are available, use module spider:
module spider LAMMPS
To learn how to load a specific module version, use module spider:
module spider LAMMPS/29Sep2021-kokkos-sm80-noMP
You will need to load all module(s) on any one of the lines below before the "LAMMPS/29Sep2021-kokkos-sm80-noMP" module is available to load.
GCC/10.2.0 CUDA/11.1.1 OpenMPI/4.0.5
Read more about toolchains.
Finally, to load LAMMPS:
module load GCC/10.2.0 CUDA/11.1.1 OpenMPI/4.0.5 LAMMPS/29Sep2021-kokkos-sm80-noMP
To find what LAMMPS versions are available, use module spider:
module spider LAMMPS
To learn how to load a specific module version, use module spider:
module spider LAMMPS/29Aug2024-CUDA-12.5.0
To load LAMMPS:
module load LAMMPS/29Aug2024-CUDA-12.5.0
To find what LAMMPS versions are available, use module spider:
module spider LAMMPS
To learn how to load a specific module version, use module spider:
module spider LAMMPS/29Aug2024_stable
To load LAMMPS:
module load LAMMPS/29Aug2024_stable
Software modules may include one of several acceleration packages:
- Software modules with Kokkos in the tag will include the Kokkos package.
- Software modules with CUDA in the tag (but not Kokkos) will include the GPU package.
- Software modules with Intel in the tag will include the GPU package.
- Other modules are CPU-only and most likely include the Kokkos package.
LAMMPS Containers
Container images built for NVIDIA GPUs can be found on the NVIDIA Container Registry
Example using singularity runtime on a compute node:
- Fetch the container image:
- Execute LAMMPS:
NVIDIA containers always include the Kokkos package.
LAMMPS Executables
The normal LAMMPS executable is called lmp. However, some variants exist:
- LAMMPS executable for CPUs is often called
lmp_mpi - LAMMPS executable for Intel hardware is often called
lmp_oneapi - On older clusters, LAMMPS executable were sometimes named
lmp_gpuorlmp_linux
If you get bash: lmp: command not found..., try one of the variants.
Usage on the Login Nodes
Please limit interactive processing to short, non-intensive usage. Use non-interactive batch jobs for resource-intensive and/or multiple-core processing. Users are requested to be responsible and courteous to other users when using software on the login nodes.
The most important processing limits here are:
* ONE HOUR of PROCESSING TIME per login session.
- EIGHT CORES per login session on the same node or (cumulatively) across all login nodes.
Anyone found violating the processing limits
will have their processes killed without warning. Repeated violation of
these limits will result in account suspension.
Note: Your login session will disconnect after
one hour of inactivity.
Using Drona Workflow Engine to run LAMMPS Jobs on the Compute Nodes
Note: The Drona LAMMPS environment is under active development and is currently only available on the ACES cluster.
Drona Workflow Engine, developed by HPRC, provides a 100% graphical interface to generate and submit LAMMPS jobs without the need to write a Slurm script yourself or even be aware of Slurm syntax and LAMMPS internals. The Drona app is available on all HPRC Portals under the Jobs tab (Screenshot) .
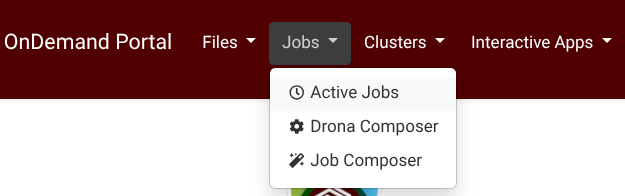{kind=link}
you will find LAMMPS in the Environments Dropdown (Screenshot). NOTE: If you don't see LAMMPS in the Environments Dropdown, you need to import it first. Click on the + sign next to the environments dropdown and select the LAMMPS environment in the pop-up window. You only need to do this once. See the import section for more information about environments.
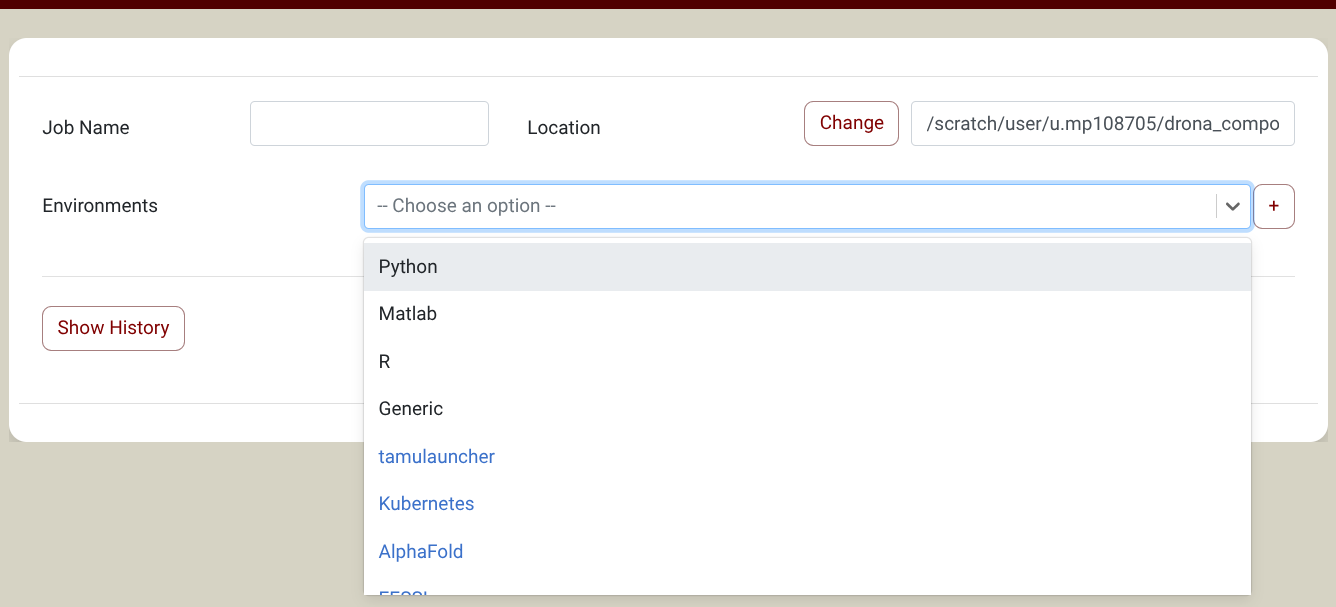{kind=link}
Once you select the LAMMPS environment, the form will expand with LAMMPS specific fields (Screenshot) to guide you in providing all the needed information. To generate the LAMMPS job files, click the Generate or Preview button. This will first show a fully editable preview screen with the generated job scripts. You are welcome to inspect the generated files and make edits if needed. To submit the job, click on the submit button, and Drona will submit the generated job on your behalf. For detailed information about Drona Workflow Engine, checkout the Drona Workflow Engine Guide
{kind=link}
If you experience any issues or have any suggestions, please get in touch with us at [email protected]
Usage on the Compute Nodes
Non-interactive batch jobs on the compute nodes allows for resource-demanding processing. Non-interactive jobs have higher limits on the number of cores, amount of memory, and runtime length.
For instructions on how to create and submit a batch job, please see the appropriate wiki page for each respective cluster:
- Grace: About Grace Batch Processing
- FASTER: About Grace Batch Processing
- ACES: About Grace Batch Processing
To submit a batch job, run:
[NetID@login ~]$sbatchjobscript
NVIDIA GPU Job Example
Example coming soon!
Example coming soon!
#!/bin/bash
## NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=lammps # Set the job name to "lammps"
#SBATCH --time=01:30:00 # Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=1 # Request 1 core
#SBATCH --nodes=1 # Request 1 node
#SBATCH --mem=40G # Request 40GB per node
#SBATCH --partition=gpu # Reqeust a node with an NVIDIA GPU
#SBATCH --gres=gpu:h100:1 # Request 1 H100 GPU per node
#SBATCH --output=lammps.%j # Send stdout/err to "lammps.[jobID]"
module load LAMMPS/29Aug2024-CUDA-12.5.0-Kokkos-4.3.1
mpirun -np 1 lmp -k on g 1 -sf kk -in inputfile
For the Kokkos package, performance is better when the number of processes is equal to the number of GPUs.
Example coming soon!
Intel GPU Job Example
Grace doesn't have Intel GPUs.
FASTER doesn't have Intel GPUs.
#!/bin/bash
# JOB SPECIFICATIONS
#SBATCH --job-name=lammps # Set the job name to "lammps"
#SBATCH --time=01:30:00 # Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=96 # Request 96 cores (see note)
#SBATCH --nodes=1 # Request 1 node
#SBATCH --mem=40G # Request 40GB per node
#SBATCH --partition=pvc # Reqeust a node with an Intel GPU
#SBATCH --gres=gpu:pvc:1 # Request 1 PVC GPU per node
#SBATCH --output=lammps.%j # Send stdout/err to "lammps.[jobID]"
module load intel-compilers/2023.2.1 impi/2021.10.0
module load LAMMPS/2Aug2023_update2-intel-2023.07
mpirun -np 24 lmp_oneapi -pk gpu 1 -sf gpu -in inputfile
For the GPU package, performance is better when the job runs alone on a node. This can be accomplished by requesting all of the 96 cores on a node, or by using the --exclusive flag.
Launch doesn't have Intel GPUs.
MPI Job Example
#!/bin/bash
## NECESSARY JOB SPECIFICATIONS
#SBATCH --job-name=lammps # Set the job name to "lammps"
#SBATCH --time=01:30:00 # Set the wall clock limit to 1hr and 30min
#SBATCH --ntasks=56 # Request 56 tasks
#SBATCH --ntasks-per-node=28 # Request 28 task per node
#SBATCH --mem=40G # Request 40GB per node
#SBATCH --output=lammps.%j # Send stdout/err to "lammps.[jobID]"
module load GCC/8.3.0 OpenMPI/3.1.4
module load LAMMPS/3Mar2020-Python-3.8.2-kokkos
mpirun lmp -in inputfile
Example coming soon!
Example coming soon!
Example coming soon!
HPRC Publications
LAMMPS using A100 on FASTER and Grace
- Developing Synthetic Applications Benchmarks on Composable Cyberinfrastructure: A Study of Scaling Molecular Dynamics Applications on GPUs
LAMMPS using H100 and PVC on ACES
- Performance of Molecular Dynamics Acceleration Strategies on Composable Cyberinfrastructure