AlphaFold
GPU Support
-
Currently AlphaFold 2 and 3 only supports NVIDIA GPUs and not Intel PVC GPUs.
-
Currently AlphaFold 2 and 3 only supports running on a single GPU
-
Use ParaFold to run AlphaFold2 On the HPRC clusters to avoid idle GPU usage.
GCATemplates
Description
AlphaFold is an AI system that predicts a protein’s 3D structure from its amino acid sequence.
Scientists can access the majority of the AlphaFold3 capabilities, for free, through the AlphaFold Server
AlphaFold3 homepage
AlphaFold2 homepage
AlphaFold3
You will need your own copy of the model parameters file which you can obtain after accepting the google agreement.
AlphaFold3 used .json input file format. There are example input .json format files in the directory /scratch/data/bio/alphafold3/examples/
Run the alphafold3jobscript utility on any HPRC cluster to generate an AlphaFold3 jobscript.
alphafold3jobscript --help
Synopsis:
alphafold3jobscript is a utility to create an AlphaFold3 job script to first run a CPU-only job for the
sequence alignment steps and then run a second GPU job for the structure prediction step.
Required:
--json_path /full/path/to/input.json # full path to your input.json file
--model_dir /full/path/to/model/dir # full path to the directory containing your af3.bin.zst model file
Optional:
--max_template_date date # format YYYY-MM-DD (default: 2025-01-01)
--num_recycles int # (default: 3)
--output_dir /full/path/to/dir # (default: $PWD/output_NAME_JOBID)
--gpu_type type # a30, h100 (default: first available)
Example Command:
alphafold3jobscript --json_path /full/path/to/my/alphafold_input.json --model_dir /full/path/to/my/model/dir/
Model Parameters File:
https://docs.google.com/forms/d/e/1FAIpQLSfWZAgo1aYk0O4MuAXZj8xRQ8DafeFJnldNOnh_13qAx2ceZw/viewform
The --gpu-type option is required for clusters that have GPUs with compute capabilities = 7.5
AlphaFold2
AlphaFold 2.3.2+ is available on Grace as a singularity container created from tacc/alphafold
/sw/hprc/sw/bio/containers/alphafold/alphafold_2.3.2.sif
The AlphaFold 2.3.2 databases are found in the following directory
/scratch/data/bio/alphafold/2.3.2
If using the singularity image, you can either run alphafold using the following command while using the VNC portal app or from within your job script.
Be sure to select a GPU when launching the VNC app.
singularity exec /sw/hprc/sw/containers/alphafold/alphafold_2.3.2.sif python /app/alphafold/run_alphafold.py --helpfull
See the Biocontainers page for additional details on how to use .sif image files.
ParaFold
ParaFold is the preferred approach to run AlphaFold on the HPRC clusters, especially ACES, since it does not hold the GPU idle for hours during the multiple sequence alignment step.
ParaFold This project is a modified version of DeepMind's AlphaFold2 to achieve high-throughput protein structure prediction.
To find what ParaFold versions are available, use module spider:
module spider ParaFold
To learn how to load a specific module version, use module spider:
module spider ParaFold/2.0-CUDA-11.7.0
You will need to load all module(s) on any one of the lines below before the "ParaFold/2.0-CUDA-11.7.0" module is available to load.
GCC/11.3.0 OpenMPI/4.1.4
Read more about toolchains.
Finally, to load ParaFold:
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.7.0
To find what ParaFold versions are available, use module spider:
module spider ParaFold
To learn how to load a specific module version, use module spider:
module spider ParaFold/2.0-CUDA-11.7.0
You will need to load all module(s) on any one of the lines below before the "ParaFold/2.0-CUDA-11.7.0" module is available to load.
GCC/11.3.0 OpenMPI/4.1.4
Read more about toolchains.
Finally, to load ParaFold:
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.7.0
To find what ParaFold versions are available, use module spider:
module spider ParaFold
To learn how to load a specific module version, use module spider:
module spider ParaFold/2.0-CUDA-11.8.0
You will need to load all module(s) on any one of the lines below before the "ParaFold/2.0-CUDA-11.8.0" module is available to load.
GCC/11.3.0 OpenMPI/4.1.4
Read more about toolchains.
Finally, to load ParaFold:
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.8.0
Example Job Scripts
ParaFold is available on Grace and is the preferred approach to running AlphaFold2 because it avoids idle GPU usage.
#!/bin/bash
#SBATCH --job-name=parafold-cpu # job name
#SBATCH --time=1-00:00:00 # max job run time dd-hh:mm:ss
#SBATCH --ntasks-per-node=1 # tasks (commands) per compute node
#SBATCH --cpus-per-task=24 # CPUs (threads) per command
#SBATCH --mem=180G # total memory per node
#SBATCH --output=stdout.%x.%j # save stdout to file
#SBATCH --error=stderr.%x.%j # save stderr to file
<<README
- AlphaFold2 manual: https://github.com/deepmind/alphafold
- ParaFold: https://github.com/Zuricho/ParallelFold
README
######### SYNOPSIS #########
# this script will run ParaFold which runs AlphaFold2 in two jobs, one for the CPU step
# and a second for the GPU steps and graph .pkl files.
# currently AlphaFold2 supports running on only one GPU
module purge
module load GCC/11.3.0 OpenMPI/4.1.4
module load ParaFold/2.0-CUDA-11.7.0
################################### VARIABLES ##################################
########## INPUTS ##########
protein_fasta='/scratch/data/bio/alphafold/example_data/1L2Y.fasta'
######## PARAMETERS ########
ALPHAFOLD_DATA_DIR=/scratch/data/bio/alphafold/2.3.2 # 3TB data already downloaded here
max_template_date=2025-1-1
model_preset=monomer_ptm # Options: monomer, monomer_casp14, monomer_ptm, multimer
# parafold does not support reduced_dbs
########## OUTPUTS #########
pf_output_dir=out_parafold_${model_preset}
################################### COMMANDS ###################################
jobstats &
# First, run CPU-only steps to get multiple sequence alignments:
run_alphafold.sh -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date -f
# Second, run GPU steps as a separate job after the first part completes successfully:
sbatch --job-name=parafold-gpu --time=2-00:00:00 --ntasks-per-node=1 --cpus-per-task=24 --mem=180G --gres=gpu:a100:1 --partition=gpu --output=stdout.%x.%j --error=stderr.%x.%j --dependency=afterok:$SLURM_JOBID<<EOF
#!/bin/bash
module purge
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.7.0
module load AlphaPickle/1.4.1
jobstats &
echo "run_alphafold.sh -g -u 0 -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date"
run_alphafold.sh -g -u 0 -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date
jobstats
EOF
jobstats
#!/bin/bash
#SBATCH --job-name=parafold-cpu # job name
#SBATCH --time=1-00:00:00 # max job run time dd-hh:mm:ss
#SBATCH --ntasks-per-node=1 # tasks (commands) per compute node
#SBATCH --cpus-per-task=32 # CPUs (threads) per command
#SBATCH --mem=125G # total memory per node
#SBATCH --output=stdout.%x.%j # save stdout to file
#SBATCH --error=stderr.%x.%j # save stderr to file
<<README
- AlphaFold2 manual: https://github.com/deepmind/alphafold
- ParaFold: https://github.com/Zuricho/ParallelFold
README
######### SYNOPSIS #########
# This script runs ParaFold which executes AlphaFold2 in two steps:
# 1. A CPU-only step for multiple sequence alignments.
# 2. A GPU step for structure prediction and graph generation.
# Note: AlphaFold2 currently supports running on only one GPU.
# Load necessary modules:
module purge
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.7.0
################################### VARIABLES ##################################
# Inputs
protein_fasta='/scratch/data/bio/alphafold/example_data/1L2Y.fasta'
# Parameters
ALPHAFOLD_DATA_DIR=/scratch/data/bio/alphafold/2.3.2 # 3TB data already downloaded here
max_template_date=2025-1-1
model_preset=monomer_ptm # Options: monomer, monomer_casp14, monomer_ptm, multimer
# Note: ParaFold does not support reduced_dbs
# Outputs
pf_output_dir=out_parafold_${model_preset}
################################### COMMANDS ###################################
# Monitor job resource usage:
jobstats &
# Step 1: Run CPU-only steps to generate multiple sequence alignments:
run_alphafold.sh -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date -f
# Step 2: Submit GPU job after successful CPU completion:
sbatch --job-name=parafold-gpu \
--time=2-00:00:00 \
--ntasks-per-node=1 \
--cpus-per-task=24 \
--mem=180G \
--gres=gpu:t4:1 \
--output=stdout.%x.%j \
--error=stderr.%x.%j \
--dependency=afterok:$SLURM_JOBID <<EOF
#!/bin/bash
module purge
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.7.0
# Monitor GPU resource usage:
jobstats &
echo "Executing GPU step:"
echo "run_alphafold.sh -g -u 0 -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date"
# Run GPU-enabled step:
run_alphafold.sh -g -u 0 -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date
jobstats
EOF
# Final resource stats:
jobstats
This is an example job script that will run the two parts of parafold. The first part is a job that runs multiple sequence alignments on a CPU only node. When that job completes successfully, the second job is submitted by the first job script. The second job will run the structure prediction part on a GPU node.
#!/bin/bash
#SBATCH --job-name=parafold-cpu # job name
#SBATCH --time=1-00:00:00 # max job run time dd-hh:mm:ss
#SBATCH --ntasks-per-node=1 # tasks (commands) per compute node
#SBATCH --cpus-per-task=48 # CPUs (threads) per command
#SBATCH --mem=244G # total memory per node
#SBATCH --output=stdout.%x.%j # save stdout to file
#SBATCH --error=stderr.%x.%j # save stderr to file
module purge
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.8.0
ALPHAFOLD_DATA_DIR=/scratch/data/bio/alphafold/2.3.2
protein_fasta='/scratch/data/bio/alphafold/example_data/1L2Y.fasta'
model_preset=monomer_ptm # monomer, monomer_casp14, monomer_ptm, multimer
max_template_date=2024-1-1
jobstats &
# First, run CPU-only steps to get multiple sequence alignments:
run_alphafold.sh -d $ALPHAFOLD_DATA_DIR -o parafold_output_dir -i $protein_fasta -p $model_preset -t $max_template_date -f
# Second, run GPU steps as a separate job after the first part completes successfully:
sbatch --job-name=parafold-gpu --time=2-00:00:00 --ntasks-per-node=1 --cpus-per-task=24 --mem=122G --gres=gpu:h100:1 --partition=gpu --output=stdout.%x.%j --error=stderr.%x.%j --dependency=afterok:$SLURM_JOBID<<EOF
#!/bin/bash
module purge
module load GCC/11.3.0 OpenMPI/4.1.4 ParaFold/2.0-CUDA-11.8.0
jobstats -i 1 &
echo "run_alphafold.sh -g -u 0 -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date"
run_alphafold.sh -g -u 0 -d $ALPHAFOLD_DATA_DIR -o $pf_output_dir -p $model_preset -i $protein_fasta -t $max_template_date
jobstats
EOF
jobstats
Using Drona Workflow Engine to run AlphaFold Jobs
Drona Workflow Engine, developed by HPRC, provides a 100% graphical interface to generate and submit AlphaFold jobs without the need to write a Slurm script yourself or even be aware of Slurm syntax and AlphaFold internals. The Drona app is available on all HPRC Portals under the Jobs tab (Screenshot) .
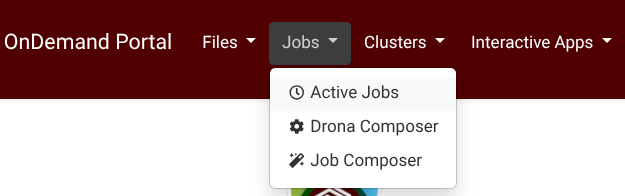{kind=link}
you will find AlphaFold in the Environments Dropdown (Screenshot). NOTE: If you don't see AlphaFold in the Environments Dropdown, you need to import it first. Click on the + sign next to the environments dropdown and select the AlphaFold environment in the pop-up window. You only need to do this once. See the import section for more information about environments.
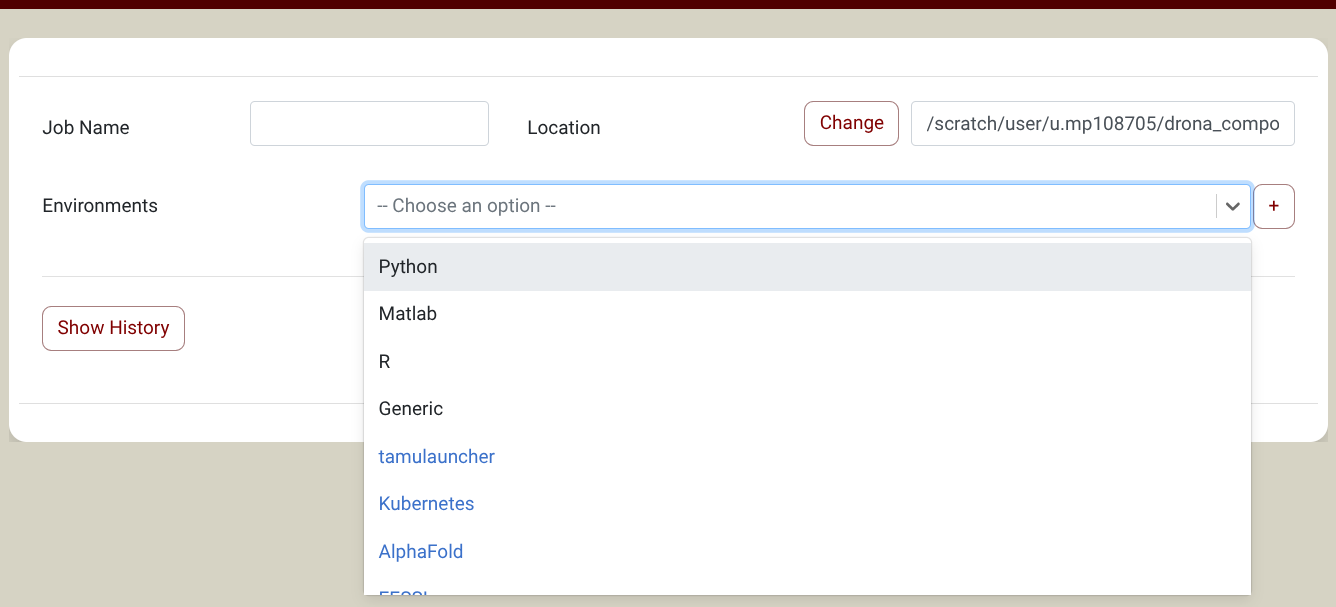{kind=link}
Once you select the AlphaFold environment, the form will expand with AlphaFold specific fields (Screenshot) to guide you in providing all the needed information. To generate the AlphaFold job files, click the Generate or Preview button. This will first show a fully editable preview screen with the generated job scripts. You are welcome to inspect the generated files and make edits if needed. To submit the job, click on the submit button, and Drona will submit the generated job on your behalf. For detailed information about Drona Workflow Engine, checkout the Drona Workflow Engine Guide
{kind=link}
If you experience any issues or have any suggestions, please get in touch with us at [email protected]
Visualize Results
AlphaPickle can be used to create graphs for visualizing each of the model .pkl files.
#!/bin/bash
#SBATCH --job-name=alphafold # job name
#SBATCH --time=2-00:00:00 # max job run time dd-hh:mm:ss
#SBATCH --ntasks-per-node=1 # tasks (commands) per compute node
#SBATCH --cpus-per-task=24 # CPUs (threads) per command
#SBATCH --mem=180G # total memory per node
#SBATCH --gres=gpu:a100:1 # request 1 A100 GPU
#SBATCH --output=stdout.%x.%j # save stdout to file
#SBATCH --error=stderr.%x.%j # save stderr to file
module purge
module load GCC/10.2.0 CUDA/11.1.1
OpenMPI/4.0.5 AlphaPickle/1.4.1
export SINGULARITYENV_TF_FORCE_UNIFIED_MEMORY=1
export SINGULARITYENV_XLA_PYTHON_CLIENT_MEM_FRACTION=4.0
DOWNLOAD_DIR=/scratch/data/bio/alphafold/2.3.2
# run gpustats in the background (&) to monitor gpu usage in order to create a graph later
gpustats &
singularity exec --nv /sw/hprc/sw/bio/containers/alphafold/alphafold_2.1.2.sif python /app/alphafold/run_alphafold.py \
--use_gpu_relax \
--data_dir=$DOWNLOAD_DIR \
--uniref90_database_path=$DOWNLOAD_DIR/uniref90/uniref90.fasta \
--mgnify_database_path=$DOWNLOAD_DIR/mgnify/mgy_clusters_2018_12.fa \
--bfd_database_path=$DOWNLOAD_DIR/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--use_gpu_relax \
--data_dir=$DOWNLOAD_DIR \
--uniref90_database_path=$DOWNLOAD_DIR/uniref90/uniref90.fasta \
--mgnify_database_path=$DOWNLOAD_DIR/mgnify/mgy_clusters_2018_12.fa \
--bfd_database_path=$DOWNLOAD_DIR/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--uniclust30_database_path=$DOWNLOAD_DIR/uniclust30/uniclust30_2021_03/UniRef30_2021_03 \
--pdb70_database_path=$DOWNLOAD_DIR/pdb70/pdb70 \
--template_mmcif_dir=$DOWNLOAD_DIR/pdb_mmcif/mmcif_files \
--obsolete_pdbs_path=$DOWNLOAD_DIR/pdb_mmcif/obsolete.dat \
--model_preset=monomer \
--max_template_date=2022-1-1 \
--db_preset=full_dbs \
--output_dir=out_alphafold \
--fasta_paths=/scratch/data/bio/alphafold/example_data/T1050.fasta
# run gpustats to create a graph of gpu usage for this job
gpustats
# run AlphaPickle to create a graph for each model .pkl file.
# Name the -od directory based on how you named --output_dir and --fasta_paths in the run_alphafold.py command
run_AlphaPickle.py -od out_alphafold/T1050
Database directory
The AlphaFold2 database directory is structured as follows: check for the latest version in /scratch/data/bio/alphafold/
/scratch/data/bio/alphafold/2.3.2/
├── bfd
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffdata
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_a3m.ffindex
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffdata
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_cs219.ffindex
│ ├── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffdata
│ └── bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt_hhm.ffindex
├── example_data
│ ├── 1L2Y.fasta
│ ├── mmcif_3geh.fa
│ ├── T1083.fasta
│ └── T1083_T1084_multimer.fasta
├── mgnify
│ └── mgy_clusters_2022_05.fa
├── params
│ ├── LICENSE
│ ├── params_model_1_multimer_v3.npz
│ ├── params_model_1.npz
│ ├── params_model_1_ptm.npz
│ ├── params_model_2_multimer_v3.npz
│ ├── params_model_2.npz
│ ├── params_model_2_ptm.npz
│ ├── params_model_3_multimer_v3.npz
│ ├── params_model_3.npz
│ ├── params_model_3_ptm.npz
│ ├── params_model_4_multimer_v3.npz
│ ├── params_model_4.npz
│ ├── params_model_4_ptm.npz
│ ├── params_model_5_multimer_v3.npz
│ ├── params_model_5.npz
│ ├── params_model_5_ptm.npz
├── pdb70
│ ├── md5sum
│ ├── pdb70_a3m.ffdata
│ ├── pdb70_a3m.ffindex
│ ├── pdb70_clu.tsv
│ ├── pdb70_cs219.ffdata
│ ├── pdb70_cs219.ffindex
│ ├── pdb70_hhm.ffdata
│ ├── pdb70_hhm.ffindex
│ └── pdb_filter.dat
├── pdb_mmcif
│ ├── mmcif_files
│ └── obsolete.dat
├── pdb_seqres
│ └── pdb_seqres.txt
├── small_bfd
│ └── bfd-first_non_consensus_sequences.fasta
├── uniprot
│ └── uniprot.fasta
├── uniref30
│ ├── UniRef30_2023_02_a3m.ffdata
│ ├── UniRef30_2023_02_a3m.ffindex
│ ├── UniRef30_2023_02_cs219.ffdata
│ ├── UniRef30_2023_02_cs219.ffindex
│ ├── UniRef30_2023_02_hhm.ffdata
│ ├── UniRef30_2023_02_hhm.ffindex
│ └── UniRef30_2023_02.md5sums
└── uniref90
└── uniref90.fasta
Usage
If using the AlphaFold2 software module, run the following for usage:
run_alphafold.py --helpfull
If using the ParaFold software module, run the following for usage:
run_alphafold.sh --help
FAQ
Q. I'm seeing the message "RuntimeError: Resource exhausted: Out of memory"
A. make sure you have the following two lines in your AlphaFold2 job script. This is not needed with ParaFold since it is automatically configured in the ParaFold software module.
export SINGULARITYENV_TF_FORCE_UNIFIED_MEMORY=1
export SINGULARITYENV_XLA_PYTHON_CLIENT_MEM_FRACTION=4.0
Q. I'm seeing the message: raise ValueError('The number of positions must match the number of atoms')
A. See this bug report
Q. How do I view the contents of a .pkl file such as result_model_2.pkl?
A. Use python to view the contents. More code needed to save to a file.
module purge
module load GCC/10.2.0 CUDA/11.1.1 OpenMPI/4.0.5 AlphaPickle/1.4.1
python
>>> import pandas as pd
>>> myresults=pd.read_pickle('/path/to/your/result_model_2.pkl')
>>> print(myresults)
Q. How do I create graphs of the .pkl model files
A. Use AlphaPickle which can be run on the login node after an AlphaFold2 job is complete since AlphaPickle takes less than a minute to complete.
module purge
module load GCC/10.2.0 CUDA/11.1.1 OpenMPI/4.0.5 AlphaPickle/1.4.1
# run AlphaPickle to create a graph for each model .pkl file.
# Name the -od directory based on how you named --output_dir and --fasta_paths in the run_alphafold.py command
run_AlphaPickle.py -od out_alphafold/T1050
Q. How can I extract ptm, iptm and ranking_confidence values from a pickle file such as result_model_2.pkl?
A. Use pickle2csv.py to extract the values for ptm, iptm and ranking_confidence when using --model_preset=monomer_ptm
If you used --model_preset=monomer then only ranking_confidence will be extracted
module purge
module load GCC/10.2.0 CUDA/11.1.1 OpenMPI/4.0.5 AlphaPickle/1.4.1
pickle2csv.py -i path/to/pickle/file/result_model_2.pkl -o output.csv
Q. Does AlphaFold run on the ACES PVC accelerators?
A. No. PVC accelerators are not supported by AlphaFold2 or AlphaFold3
Citation
- If you use an AlphaFold prediction in your work, please cite the following papers:
Jumper, J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021).
Varadi, M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2021).
- In addition, if you use the AlphaFold-Multimer mode, please cite:
Evans, R et al. Protein complex prediction with AlphaFold-Multimer, doi.org/10.1101/2021.10.04.463034
- In addition, if you use ParaFold, please cite:
Bozitao Zhong, Xiaoming Su, Minhua Wen, Sichen Zuo, Liang Hong, James Lin. ParaFold: Paralleling AlphaFold for Large-Scale Predictions. 2021. arXiv:2111.06340. doi.org/10.48550/arXiv.2111.06340
- In addition, if you use AlphaPickle, please cite:
Arnold, M. J. (2021) AlphaPickle, doi.org/10.5281/zenodo.5708709